Abstract: The rare disease of chronic infantile neurological cutaneous and articular (CINCA) syndrome, is caused by the over-secretion of interleukin (IL)-1β due to a gain-of-function NLRP3 gene mutation in the autosomal chromosome which often involves in eyes. In this report, we studied a 9-year-old girl with CINCA. The eyes were also involved and presented bilateral papilledema. Genetic testing revealed that the symptoms were caused by a novel gene mutation site (c.913G>A, p. D305N) in conservative domain exon-3 of NLRP3 which is gain-function gene of CINCA. The patient had the characteristic facial features, frontal fossa and saddle nose, manifested the generalized urticaria-like skin rash at two weeks after birth, periodic fever 6 months after birth, sensorineural deafness at 7 years old, and bilateral papilledema, aseptic meningitis and knee arthropathy at 9 years old. White cell counts, C-reactive protein increased and intracranial pressure raised to 300 mmH2O. The meningeal thickening enhanced by gadolinium in magnetic resonance imaging (MRI). Based on clinical features and genetic test, the girl was diagnosed bilateral papilledema secondary to CINCA and administered prednisone and lowered intracranial pressure medicine to resolve symptoms. With 3-year follow-up, patient had no inflammatory flare-up with visual acuity improvement. The finding of novel genetic mutation site (p. D305N) in NLRP3 gene expanded genotype spectrum associated with CINCA. This case also expanded the cause spectrum of papilledema and it highlighted systemic disease history for patients with bilateral papilledema.
Papilledema or optic disc swelling is the most common signs of neuro-ophthalmology diseases. Papilledema usually is caused by intracranial hypertension and optic disc swelling often results from the optic nerve or optic disc inflammation. They are apparently same and difficulty to distinguish at the onset of neuro-ophthalmology diseases. Their most common causes are optic neuritis (ON), about 73% pediatric ON presented with optic disc swelling (1). The second major causes are intracranial hypertension secondary to cranial diseases. However, bilateral papilledema seconded by chronic infantile neurological cutaneous and articular (CINCA) syndrome is very rare. In this article we will present a bilateral papilledema case caused by CINCA with a novel a novel (p. D305N) mutation site in NLRP3 (2). CINCA is a rare disease of CINCA syndrome, is the most severe phenotype of cryopyrin-associated periodic syndrome (CAPS). The syndrome is caused by the over-secretion of interleukin (IL)-1β due to a gain-of-function NLRP3 gene mutation in the autosomal chromosome (3). We present the following article in accordance with the CARE reporting checklist (available at http://dx.doi.org/10.21037/aes-20-140).
This study was performed with the approval of the ethics committee of Beijing Children’s Hospital, Capital Medical University, and the study adhered to the Declaration of Helsinki (the 2013 revision), the guidelines of the International Conference on Harmonisation of Good Clinical Practice and applicable Chinese laws. Guardian’s consent about patient’s data presentation was obtained.
A 9-year-old girl complained suddenly of vision loss in both eyes for more than 1 month with headache, and nausea for a week.
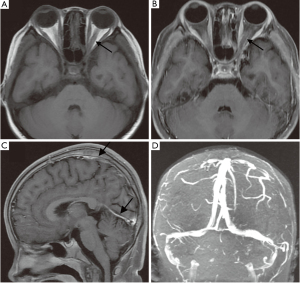
Physical examinations (PE) revealed the following: vision acuity (VA) of 10/40 in the right eye and FC/20 cm in the left eye relative afferent pupil defect (RAPD) was positive in the left eye; and optic disc swollen and pallor was found in both eyes (Figure 1). The girl had a face characterized by frontal fossa and saddle nose which were distinct from her parents. The brain magnetic resonance imaging (MRI) scanning excluded tumors and other cerebral diseases. Then the girl was primarily diagnosed as bilateral ON without VA improvement and optic disc swollen continued after pulse steroid therapy followed by tapered of dose for 1 month.

Spinal lumbar puncture showed intracranial hypertension (300 mmH2O) and cerebral spinal fluid (CSF) tests showed: white cell count: 46×106/L↑; protein: 609.3 mg/L↑; glucose: 2.4 mmol/L and chloride: 121.4 mmol/L. The later brain MRI scanning demonstrated thickened meningeal with blurred rim in parietal and occipital lobes, and enhanced by gadolinium. The orbit MRI scanning demonstrated thickening of the left optic nerve in orbit, with slight enhancement remaining by gadolinium in orbit segment at 1 month after ON onset (Figure 2). The brain magnetic resonance venography (MRV) and magnetic resonance angiography (MRA) were normal. A week ago, the girl presented headache, vomiting and stiff neck. She was considered to be suffering from meningitis.
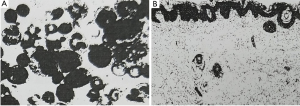
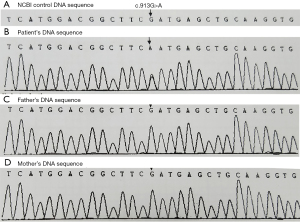
The girl suffered an urticaria-like skin rash two weeks after birth and periodic fever (38–40 °C) from 6 months after birth. The white cell count ranged from 16×109/L to 21×109/L, and C-reactive protein (CRP) ranged 8–25 mg/L and erythrocyte sedimentation rate (ESR) were normal. All auto-immune, rheumatoid, endocrine and tumor biomarkers serum tests showed negative results. The biopsy of skin tissue affected by rash demonstrated that the skin tissue was infiltrated by neutrophil granulocytes, lymphocytes and some eosinophil granulocytes (Figure 3). The bone marrow biopsy showed normal results. The steroid treatment eased the flare-up. The fever and urticaria-like skin rash continued. At age 7, she suffered sensorineural deafness. The bilateral keen joints suffered arthropathy in X-ray examinations. The peripheral blood was genetically tested with the Illumina Hiseq sequencer, and her parents by the Sanger sequencing technique. The results displayed that there was a novel genetic mutation (c.913G>A) in exon 3 in the NLRP3 gene in patients, which miscoded aspartic acid as asparagine at the 305th site (p. D305N) in the NLRP3 protein chain with her parents normal (Figure 4).
According to suddenly bilateral visual loss, optic swelling, meningeal thickening enhanced by gadolinium in MRI scanning, and systemic manifestations, the girl was diagnosed bilateral papilledema complicated with CINCA. Then, she accepted steroid therapy of methylprednisolone 20 mg/kg/d taken intravenously for 3 days and prednisolone 2 mg/kg/d taken orally and tapered by 5 mg per month, combining with reduction of intracranial pressure with 20% mannitol solution intravenously (5 mL/kg, twice a day). After 1 month, all the symptoms resolved and papilledema disappeared and began to atrophy. Within 3-year follow-up, disease flares were extinct, and near VA was sustained at 40/50 in the right eye and 10/100 in the left eye.
For this CINCA patient, there were some main PE and characteristic findings for diagnosis of CINCA. The VA tests showed severe visual loss in both eyes and the ocular fundus examination displayed bilateral optic disc swelling with pallor which implied long-term intracranial hypertension. Cranial meningeal thickness and enhanced by gadolinium in MRI scanning which prompted meningitis. Hearing loss was detected by audio acuity tests and arthropathy in limbs presented in X-ray scanning. Peripheral blood tests results supported the inflammatory activity such as white cells count and CRP increase, and ESR speeded, with auto-immune serum tests including antinuclear antibody (ANA), anti-double-stranded DNA antibody (dsDNA), anti-extractable nuclear antigen (ENA) antibodies spectrum negativity. All above PE with feature findings could provide potential evidences to diagnose CINCA. Most important, genetic screen that found pathogenetic mutation in the NLRP3, was the potential supporting for diagnosis of CINCA.
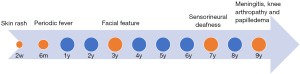
The current papilloedema and meningitis outbreaks were only a brief episode of the whole process of CINCA. Actually, the CINCA initial onset was traced back to an urticaria-like skin rash two weeks after birth and periodic fever six months after birth, followed by facial outline slowly changes from the normal to characteristic CINCA face frontal fossa and saddle nose since 3 years old, and sensorineural deafness and arthropathy at 7 years old, then the ON and meningitis outbreaks at 9 years old. Finally, all the serial clinical manifestations of neurological, cutaneous and articular of CINCA were exhibited sequentially (Figure 5).
CINCA is a systemic rare hereditary disease which frequently affected multi-organs. For the periodic fever patients, CINCA should be considered. X-ray for arthropathy, audio acuity, ocular examinations for neuropathy and brain MRI scan for meningitis were essential PE. Laboratory tests including abnormal white cell count, CRP, ESR and negative serum ANA were also vital to diagnose CINCA. The genetic screen for pathogenetic finding was potential supporting to diagnose of CINCA.
For diagnostic challenges, in the early stage of diseases characteristic clinical manifestations didn’t occur fully and it was difficult to distinguish with other infectious diseases. Due to CINCA rarity to seen, few doctors could think of it. Even if considered of CINCA, due to financial burden and lack of laboratory devices, genetic tests were not available in primary hospitals. Therefore, the diagnosis was not confirmed until multi-organs had affected in the late stage of diseases.
The girl finally was diagnosed as papilledema complicated to CINCA. The papilledema was secondary to in cranial pressure caused by meningitis. The systemic disease of CINCA diagnosed based on the clinical features and genetic test which was distinct from infectious diseases by insufficient bacterial treatment and from auto-immune disease such as lupus erythematosus and rheumatic arthritis by negativity of serum ANA.
The last episode of inflammatory flare-up occurred with multi-organs affected and patient needed life-span anti-IL-1 agent therapy. Due to huge financial burden and unavailable of IL-1 blockage, she accepted steroid therapy combining with reduction of intracranial pressure with hyperosmotic solution. Near VA was sustained at 40/50 in the right eye and 10/100 in the left eye without obvious flare-up in 3-year follow-up. However, the exophthalmos slightly worsened due to the orbit bone injury.
In order to reduce disabilities or organ function failures, the IL-1 blockage agents’ treatment should be lifelong and should be given before organs sustain permanent injuries. Steroid therapy also was a sufficient treatment to prevent deterioration of diseases.
Currently, the IL-blockage agents available were listed in Table 1.

In order to reduce disabilities or organ function failures, the disease’s response to therapy should be measured, and inflammation in multiple organs, except bones, should be monitored to confirm the flare extinction. The fetus genetic test can prevent CINCA occurrence caused by germline genetic mutation but not for somatic genetic mutations NLRP3.
Blood tests reflecting inflammatory activity including white cell count, IL-1, CRP and ESR should be tested at regular intervals to inspect inflammatory flare-ups.
In China, the adherence of IL-1 blockage agents therapy was very poor because of huge financial burden, and steroid therapy was available.
For IL-1 blockage agents therapy, the allergic reaction and insufficiency after long-term usage were concerned. As for steroid therapy, the development delay, metabolism disorder with calcium loss were main side effects.
In this case report, the bilateral papilledema complicated to CINCA caused by a novel genetic mutation site (c.913G>A, p. D305N) in NLRP3 gene was first reported. The finding of novel genetic mutation expanded genotype spectrum associated with CINCA. This case highlighted the importance of systemic disease history to bilateral papilledema. There were some limitations to weak the case report strengths. Due to retrospective studying of the 9-year history, some clinical information was inadequate or second-hand such as histopathologic images. The facial features could not be presented because fail to obtain permission of child guardian and it is very regretful to fail to have serum IL-1 assay. The response to IL-1 blockage agents’ treatment was not reported due to IL-1 unavailable and gene-function study was not preformed.
CINCA syndrome is a rare autoinflammatory disease identified by Prieur et al. in 1987 (4), also called neonatal-onset multisystem inflammatory disease (NOMID). CINCA/NOMID is the most severe type of CAPS, which also includes the phenotypes for mild familial cold autoinflammatory syndrome (FCAS) and intermedia severe Muckle-Wells syndrome (MWS) (5). Its prevalence was approximately 1–2 cases per 100,000 to 360,000 people in the United States and France, respectively (6,7). However, in China, it kept with lower prevalence worldwide, probably due to that it has not been recognized as widespread still now (8).
CINCA/NOMID usually has neonatal-onset and involves cutaneous, neurological and articular multiple systems. Levy et al. (9) revealed that fever, rash and musculoskeletal involvement were observed in all three phenotypes. Generally, CINCA/NOMID was onset from the first days of life with urticarial-like rash, followed by periodic fever and elevation of acute phase reactants (5). As for this case, the girl had skin urticarial-like rash within two weeks of birth and suffered periodic fever at 6 months, in accordance with Levy’s study. CINCA/NOMID, as well as other autoinflammatory diseases, presents serum auto-immune antibodies negative, such as ANA, anti-dsDNA, anti-ENA spectrum that because auto-inflammatory injury is mainly caused by mononuclear macrophage other than T or B cells caused by antibody or complement in autoimmune diseases (10-12). Moreover, CINCA/NOMID usually presents frontal bossing, large cephalic perimeter and saddle-back nose (5), as in this case. The girl’s clinical manifestations were almost completely compatible with the features of CINCA/NOMID.
Levy’s study showed that 71% of CAPS patients had affected eyes (9). Anterior uveitis incidence accounted for 29%, and optic disc abnormalities appropriation was about 84%, mainly presenting as papilledema and optic nerve atrophy which frequently caused by aseptic meningitis that was same as this case (13).
CINCA/NOMID is a monogenic autoinflammatory disease with gain-of-function genetic mutation in the NLRP3 gene that encodes the NLRP3 protein, also called cryopyrin. These mutations caused IL-1 overexpression and triggered the insult of autoinflammatory. Once triggered, NLRP3 interacts with other intracellular proteins and formed NLRP3 inflammasome, a key player in IL-1β pathway activation which triggers a cascade of homeostatic tissue and an extracellular oligomeric complex persistent inflammatory response (5). Approximately 50–69.2% of CINCA/NOMID patients were detected mutations in the NLRP3 gene (14,15). Up to now, there is a total of 210 variants of NLRP3 genetic mutations confirmed to be responsible for the mild-to-severe phenotypes of CAPS (http://fmf.igh.cnrs.fr/ISSAID/infevers/). Seven common NLRP3 variants accounted for 78% of the patients, with 24 rare variants in 27 of the cases. Rare NLRP3 variants tended towards earlier onset and more severity of neurological complications (9). In this case, c.913G>A, p. D305N in NLRP3 was a novel pathogenic gene mutation site that has not been previously reported which caused the severity CAP. The most similar genotype of C.914T>C in exon 3 of NLRP3, which miscoded the amino acid of 305 (L305P) caused the mild phenotype of CAP (http://fmf.igh.cnrs.fr/ISSAID/infevers/). As for therapy of CAPS, the IL-1 blocking agents are the first-line medication treatment (16,17).
In conclusion, with the observation of this case, a novel genetic mutation site of (c.913G>A, p. D305N) in NLRP3 gene for CINCA/NOMID was discovered which fulfilled the genetic database of CAPS. This result should be re-proven by gene-protein function and have serum IL-1 assay for CAPS patients in future. Moreover, this result also implies that when ON patients have no response to steroid treatment, neurological examinations and systemic medical history should be considered.









